Research in the Castro’s group
Our research is focused on identification and characterization of signaling mechanisms that control cell processes, such as growth and proliferation, and how deregulation of these mechanisms contributes to cancer. There are three main ongoing projects:
Interrupted Rheb-to-AMPK feedback signaling with cytoplasmic p27 retention: mechanisms and biological consequences (funded by FONDECYT Regular/CONICYT 1120923)
A complex of two tumor suppressor proteins, known as TSC1/2 tumor suppressor complex is the main controller of cell growth. The central role of this complex in cell growth is revealed by its inactivation and contribution to the development of Tuberous Sclerosis Complex (TSC) disease, a hamartoma syndrome characterized by development of benign tumors in several organs. In addition, inactivation of TSC1/2 occurs downstream of several pathways known to be deregulated in other hamartoma syndromes and cancer. The primary consequence of loss of TSC1/2 is the activation of the small GTPase Rheb, which in turn activates mTORC1, a kinase involved in promoting cell growth. We previously demonstrated that TSC2 functions as a GTPase-activating protein (GAP) for the inactivation of Rheb and thereby mTORC1, indicating that Rheb is the molecular link between TSC1/2 and mTORC1. Consistent with this, we recently showed that activated mTORC1 is reversed by depletion of Rheb in Tsc2-null cells, and Rheb deficiency reduced their tumorigenic potential in nude mice. In addition, we demonstrated that Rheb, independently of its regulation of mTORC1, induces cell proliferation through inhibition of the cell cycle inhibitor p27Kip1 (p27). However, how Rheb regulates p27 remains elusive. In correlation with the regulation of p27, we found that Rheb also induces mTORC1-independent AMPK activation, which is known to phosphorylate p27 and, consequently, decreases p27 nuclear accumulation and increases cytoplasmic retention of p27. There is evidence suggesting that AMPK activation and/or cytoplasmic p27 retention contribute to autophagy-mediated tumor cell survival under cancer-related stresses (see proposed model below). The general goal of this project is to identify the mechanism by which aberrant Rheb activation affects AMPK/p27 function, and to gain insights into the potential biological consequences of this regulation.
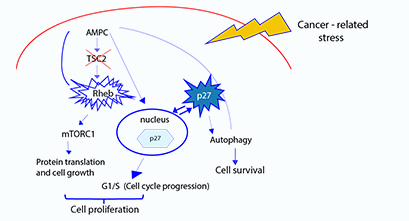
Model of proposed mechanism of Rheb regulation of p27 function and biological consequences of the mTORC1-independent Rheb functions. In the absence of TSC2 activity, aberrant Rheb activation induces mTORC1 and AMPK activation. We propose that Rheb-induced AMPK activation phosphorylates p27 resulting in decrease of nuclear p27 and
Molecular Mechanisms of Cancer Cell Resistance to Metabolic Stress: the role of NUAK1 signaling. (funded by FONDECYT Regular/CONICYT 1160731)
Cancer is a devastating disease that develops through accumulation of molecular alterations that change the genetic program driving the behavior of cells. Although there has been a significant advance in identifying some of these alterations, it is still poorly defined how cancer cells adapt to different microenvironments. In particular, it is still unclear how cancer cells survive to metabolic stress conditions caused by highly proliferative cells creating a tumor mass with a microenvironment initially far from blood vessels, resulting in shortage supply of nutrients and/or oxygen (see figure below). NUAK1 is a serine/threonine kinase that has been involved in cancer cell tolerance to metabolic stress. However, the mechanism of NUAK1 function on cancer cell survival and how this function affects tumor progression are poorly understood. In addition, it is unclear how NUAK1 is regulated during metabolic stress. Evidence indicates that NUAK1 promotes maintenance of intracellular ATP levels and cell survival under metabolic stress in a p53–independent manner. This is of particular relevance because p53 is the most frequently inactivated gene in cancer. Regarding NUAK1 regulation, we found that phosphorylation of NUAK1 increases during metabolic stress. Thus, the general goal of this project is to identify molecular mechanisms involved in NUAK1 regulation and function on cancer cell survival in response to metabolic stress.
Regulation of protein synthesis during cellular stress.
Translation of mRNA is the basic phenomenon of life that allows protein synthesis, relying in a constant expend of energy. Translational control is crucial for the correct physiological function of an organism, and is frequently disrupted in cancer cells. This phenomenon is acutely controlled by several signaling pathways, sustaining proper cell function at different conditions. Global inhibition of translation is a common response of eukaryotic cells to stressful conditions like temperature fluctuations, UV light and oxidative stress. This adaptive response allows selective and efficient synthesis of proteins relevant to the unfavorable conditions. Actively translated mRNAs form polysomes, ribonucleoprotein (RNP) complexes in which several ribosomes synthesize protein from the same mRNA molecule. However, when global translation is inhibited, stress granules (SGs) are formed. SGs are membrane-free cytoplasmic foci where components of the small ribosome subunits, untranslated mRNAs and initiation factors are sequestered and maintained inactive during cellular stress. Using different genetic, biochemical and imaging approaches, we are currently studying translational control by DISC1, a protein encoded from a gene whose mutations predispose humans to suffer schizophrenia and other mental disorders. There is evidence suggesting that DISC1 affects protein synthesis during oxidative stress, a function that could explain DISC1 association with mental disorders. In addition, DISC1 has been associated to the formation of SGs. Thus, our goal is to understand how DISC1 is involved in translational control during stress conditions.
Research in the Pincheira group
SUMMARY
Cancer is the second cause of death in Chile so it becomes relevant for our society to advance in the knowledge of this disease. What external and / or internal factors contribute to the generation of cancer? How does a benign tumor become malignant, or a primary tumor metastatic? Why do treatments fail? Why treatments work in some patients and not in others? These are some of the questions and challenges that make scientific research necessary. The long-term goal of our research is to advance in the knowledge of cancer biology, to identify participants, to know how they function and how they are related to each other. More specifically we are investigating the function of a protein called SALL2, whose levels of expression are altered in many cancers, that is compared to a normal person SALL2 levels are increased or decreased in the cancer patient. Why and how this occur, and what is the significance of this alteration in disease are some of our current questions.
WHO is SALL2?
Regulation of gene expression by transcription factors is one of the most important mechanisms governing cellular functions. Thus, it is not surprising that alteration of transcription factor function is a frequent cause of human diseases. Our laboratory has focused attention in a transcription factor named SALL2. SALL2 is a conserved member of the SAL/SPAL family of transcription factors involved in development. The gene and protein structures are shown below (from our recent review “Hermosilla V et al, Carcinogenesis, 2017”)
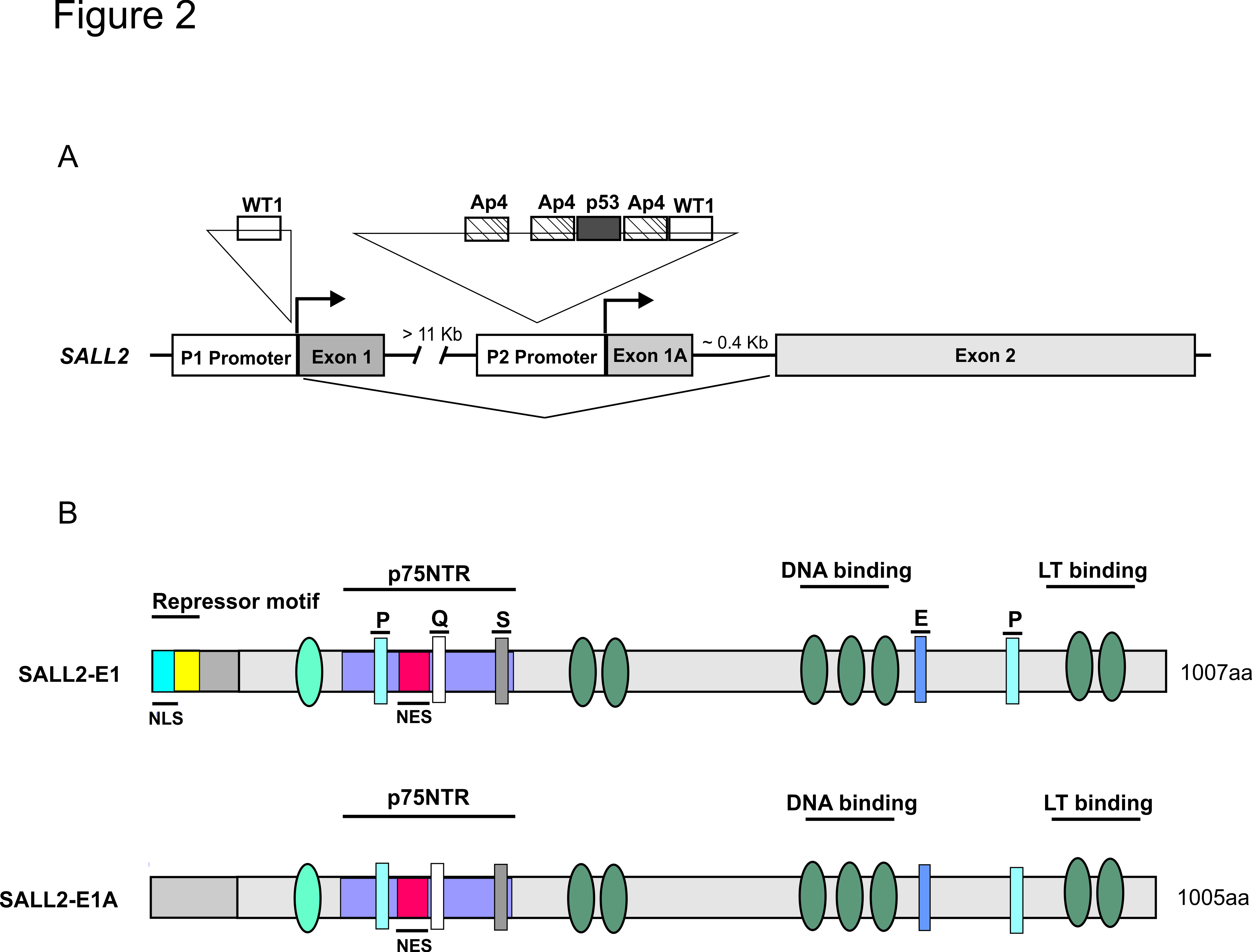
SALL2 gene and isoforms structure. (A) Human SALL2 gene located in chromosome 14 consists of exon E1, E1A and E2, and contains two alternative promoters; P1 and P2. The consensus sites for WT1, AP4 and p53 are represented by rectangles in the P1 and/or P2 promoters. (B) Human SALL2 encodes two protein isoforms, SALL2 E1 (1007 AA) and SALL2 E1A (1005 AA) as result of alternative promoters P1 and P2, respectively. Common to both SALL2 isoforms are the zinc finger domains represented by ovals and the P, Q, S and E- rich motifs represented by rectangles. Both isoforms contain the p75NTR interaction domain, DNA binding and transactivation (LT binding) domains and a putative nuclear export sequence (NES). SALL2-E1 contains a repressor domain and a putative nuclear localization signal (NLS) not present in SALL2-E1A.
WHY SALL2 is interesting?
SALL2 is a poorly characterized transcription factor that in humans has been associated with renal dysfunction, coloboma (a congenital eye disease), neurological disorders such as Alzheimer and Autism, but mainly with cancer. Unfortunately, SALL2 normal functions and the identity of the genes that it regulates are largely unknown.
SALL2 gene contains two promoters (P1 and P2) that can originate two protein isoforms; these isoforms could account for different cellular functions and might be differentially involved in disease. However, there is scarce information about SALL2 isoforms regulation, targets and function.
SALL2 is deregulated in cancers, but data is confusing; in some cancers SALL2 is found upregulated (synovial sarcoma, oral cancer, glioblastoma, wilms tumor, testicular cancer) while in others it is absent (ovarian cancer, breast cancer, leukemia). To understand SALL2 deregulation we need first to understand how SALL2 is normally regulated.
A few studies, including ours have associated SALL2 with key cancer-driver genes such as p53 and Myc. However, more studies are needed to understand and validate the functional relationship between SALL2 and these key cancer-related proteins.
It is proposed that SALL2 has tumor suppressor activity, mediated by its pro-apoptotic and cell cycle regulatory functions. In addition, SALL2 seems to play a role in cellular stemness. Several studies have associated SALL2 with stemness based on its expression levels, and its potential to interact with—or to be regulated by—key transcription factors for pluripotency maintenance. See model below (from Hermosilla V et al. Carcinogenesis 2017)
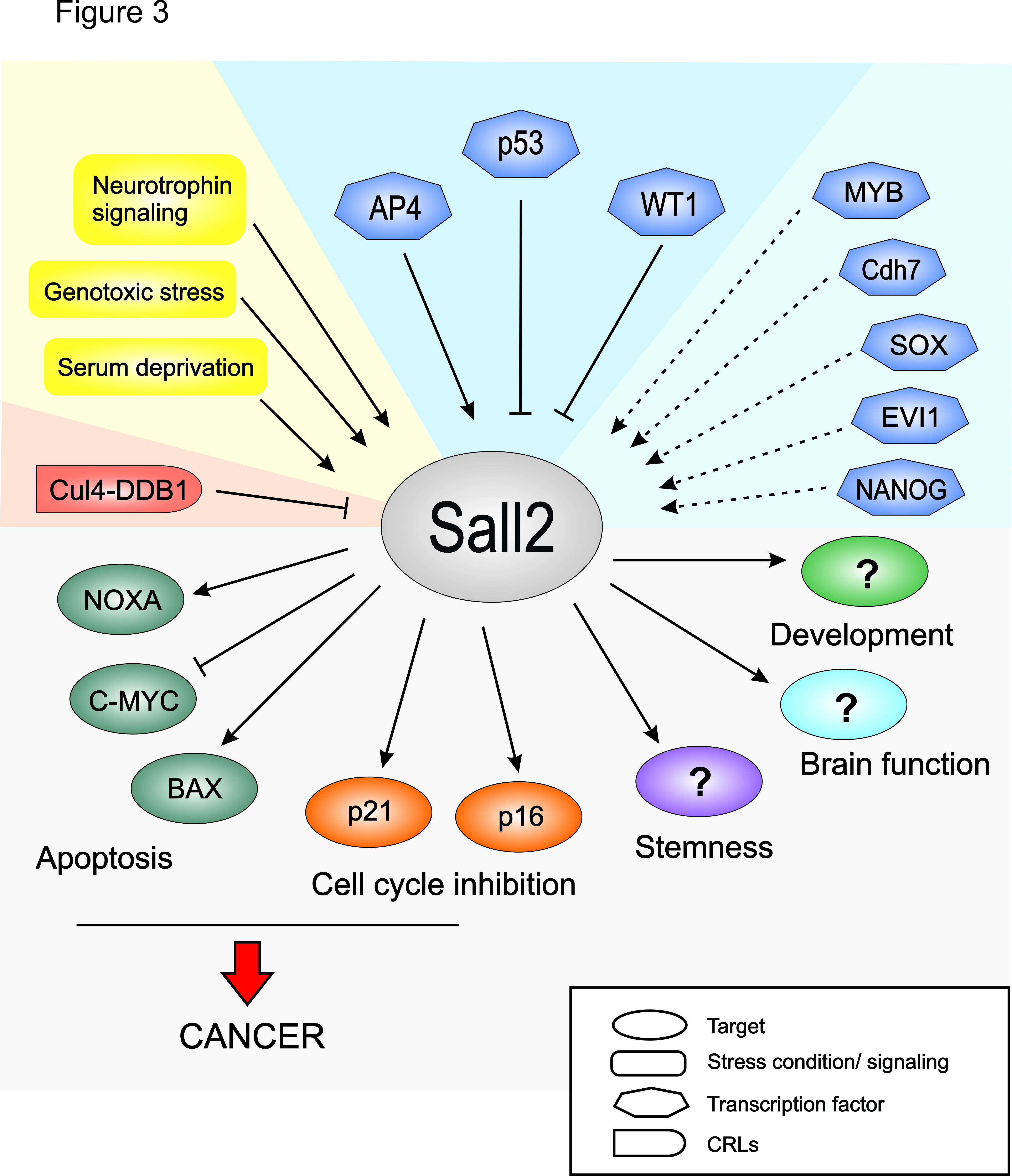
Model of SALL2 regulation and function. Stress conditions and neurotrophin signaling impact on SALL2 transcription factor activity. Depending on the cellular condition, SALL2 is transcriptionally regulated by factors such as p53, WT1 and AP4. Dashed lines indicate other putative transcriptional regulators of SALL2. On the other hand, Cul4-DDB1 negatively regulates SALL2 protein stability under certain cellular conditions. SALL2 regulates cell proliferation and apoptosis by targeting p21, p16, MYC, BAX and NOXA. In addition, SALL2 is involved in development, stemness and brain functions through unidentified targets. Deregulation of SALL2 and SALL2-dependent targets are implicated in tumorigenesis and the resistance of cancer cells to chemotherapy.
Some of our findings:
1. - SALL2 is a novel transcription factor for neurotrophin receptors and plays a role in neuronal function (Pincheira et al. EMBO J, 2009).
A model that illustrates our present understanding of Sall2 as a mediator of neurotrophin and neurotrophin receptor signaling. Sall2 constitutively associates with p75NTR (A). NGF dissociates p75NTR/Sall2 complexes and, through a TrkA-dependent process, induces translocation of Sall2 into the nucleus (B). Nuclear Sall2 plays a role in three intimately related processes that are hallmarks of the development of the neuronal phenotype: induction of p21, cell-cycle arrest, and neurite outgrowth (C).
2.- Relationship between p53 and SALL2 under genotoxic stress
We demonstrate that under genotoxic stress the p53 tumor suppressor negatively regulates SALL2 expression and activity, while mutant p53 forms commonly found in cancers do not affect SALL2 expression (Farkas C et al, PLOS 2013). However, SALL2 regulation under genotoxic stress is dynamic. At early times of stress, SALL2 is downregulated by p53, but under extended stress SALL2 levels increases in a p53-independent manner (Escobar et al, 2015). Our studies indicate that under extended stress SALL2 and p53 synergize, increasing levels of pro-apoptotic genes and promoting apoptosis .
3.- SALL2 is a stress-dependent inducible molecule
Cellular stress responses are an integral part of normal physiology to either ensure cell survival or alternatively to eliminate damaged or unwanted cells. Aberrant cellular stress responses are tightly linked to many human diseases. In particular, a change in the rate of cell death is considered to be one of the hallmarks of cancer.
SALL2 expression is regulated under stress conditions, including serum deprivation and DNA damage caused by genotoxic stress. We have shown that under genotoxic stress- induced by chemotherapy and/or radiation- Sall2 acts as a pro-apoptotic factor, independently of p53. These finding indicate that SALL2 expression is relevant for the response to cytotoxic therapies.
RELATED PROJECTS:
Regular Fondecyt 1110821 (PI) “ Characterization of Sall2 transcription factor and its relatioship to p53 tumor suppressor” (2011-2014).
Regular Fondecyt 1151031(PI) “Regulation and function of the Sall2 transcription factor during cellular stress.” (2015-2018)
Fondecyt postdoctorado 3160129 (PI. Dr. Matías Hepp) “Identificación de los genes blanco isoforma-específicos del factor de transcripción Sall2 en respuesta a estrés genotóxico”. (2016-2018)
Molecular biology techniques for cloning, site directed mutagenesis, and for gain/loss of function studies using overexpresssion, siRNA, shRNA and CRISP-Cas9 technology.
Protein-related techniques that include protein bioinformatics, Immunoprecipitation, protein fractionation, protein purification, kinase assays, Small GETPases activation assays, western blot, immunohistochemistry, Immunofluorescence and proteomics.
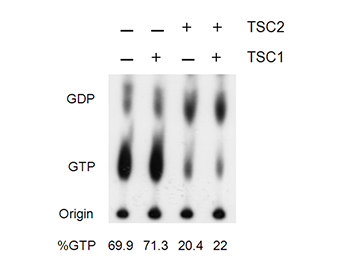
Figure shows that TSC2 decreases Rheb GTPase activity. From Castro AF et al. JBC, 2003
RNA-related techniques that include RNA isolation, RT-PCR, qPCR, RNA seq (by collaboration).
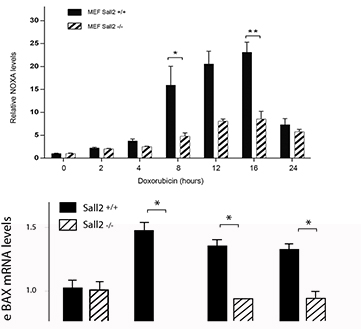
Figure shows relative NOXA mRNA expression from Sall2 wild type (Sall2 +/+) and Sall2 deficient (Sall2 -/-) primary fibroblasts treated with doxorubicin. From Escobar D, Cell Death and Disease, 2015
Transcriptional regulation techniques including gene bioinformatics, reporter assays, EMSA, ChIP.
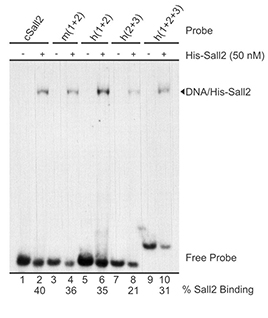
Figure shows in vitro binding of SALL2 to various regions of NOXA promoter. From Escobar et al. PLosOne, 2015.
Functional studies, we investigate the involvement of our “favorite” molecular target in the “hallmarks of cancer” by assaying various cellular functions under different stress conditions such as genotoxic and metabolic stress. These include:
Cell proliferation, cell cycle, apoptosis, cell survival, autophagy, migration, invasion, etc.
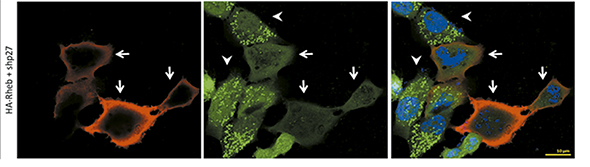
Confocal microscopy figure: Green dots show autophagy (LC3 II) in Rheb-positive cells (red). From Campos T. Molecular Carcinogenesis, 2016
Model of study: as a biological model we use several different cancer cell lines, primary cells and mice models.
Hermosilla VE, Hepp MI, Escobar D, Farlas C, Riffo EN, Castro AF, Pincheira R. Developmental SALL2 transcription factor: A new player in cancer. https://goo.gl/ZrgFrT
Campos T, Ziehe J, Palma M, Escobar D, Tapia JC, Pincheira R, Castro AF. Rheb promotes cancer cell survival through p27Kip1-dependent activation of autophagy. https://goo.gl/MCbICh
Armijo ME, Campos T, Fuentes-Villalobos F, Palma ME, Pincheira R, Castro AF. Rheb signaling and tumorigenesis: mTORC1 and new horizons. https://goo.gl/hd29ZQ
Campos T, Ziehe J, Fuentes-Villalobos F, Riquelme O, Peña D, Troncoso R, Lavandero S, Morin V, Pincheira R, Castro AF. Rapamycin requires AMPK activity and p27 expression for promoting autophagy-dependent Tsc2-null cell survival. https://goo.gl/jcJLrB
Armijo ME, Campos T, Fuentes-Villalobos F, Palma ME, Pincheira R, Castro AF. Rheb signaling and tumorigenesis:mTORC1 and new horizons. https://goo.gl/15ELuA
Escobar D, Hepp MI, Farkas C, Campos T, Sodir NM, Morales M, Álvarez CI, Swigart L, Evan GI, Gutiérrez JL, Nishinakamura R, Castro AF, Pincheira R. Sall2 is required for proapoptotic Noxa expression and genotoxic stress-induced apoptosis by dexorubicin. https://goo.gl/tW0dBg
Campos T, Ziehe J, Palma M, Escobar D, Tapia JC, Pincheira R, Castro AF. Rheb promotes cancer cell survival through p27Kip1-dependent activation of autophagy. https://goo.gl/H9quny
Farkas C, Martins CP, Escobar D, Hepp MI, Castro AF, Evan G, Gutiérrez JL, Warren R, Donner DB, Pincheira R. Wild type p53 transcriptionally represses the SALL2 transcription factor under genotoxic stress. https://goo.gl/Jm8ixF
Farkas C, Martins CP, Escobar D, Hepp MI, Castro AF, Evan G, Gutiérrez JL, Warren R, Donner DB, Pincheira R. Wild type p53 transcriptionally represses the SALL2 transcription factor under genotoxic stress. https://goo.gl/R0GLzC
Castro AF, Campos T, Babcock JT, Armijo ME, Martínez-Conde A, Pincheira R, Quilliam LA. M-Ras induces Ral and JNK activation to regulate MEK/ERK-independent gene expression in MCF-7 breast cancer cells. https://goo.gl/9x1SN4
Castro AF, Campos T, Babcock JT, Armijo ME, Martínez-Conde A, Pincheira R, Quilliam LA. M-Ras induces Ral and JNK activation to regulate MEK/ERK-independent gene expression in MCF-7 breast cancer cells. https://goo.gl/I8jUxr
Lacher MD, Pincheira RJ, Castro AF. Consequences of interrupted Rheb-to-AMPK feedback signaling in tuberous sclerosis complex and cancer. https://goo.gl/v6oFGZ
Lacher MD, Pincheira RJ, Castro AF. Consequences of interrupted Rheb-to-AMPK feedback signaling in tuberous sclerosis complex and cancer. https://goo.gl/IGWneV
Lacher MD, Pincheira R, Zhu Z, Camoretti-Mercado B, Matli M, Warren RS, Castro AF. Rheb activates AMPK and reduces p27Kip1 levels in Tsc2-null cells via mTORC1-independent mechanisms: implications for cell proliferation and tumorigenesis. https://goo.gl/h9mSv6
Lacher MD, Pincheira R, Zhu Z, Camoretti-Mercado B, Matli M, Warren RS, Castro AF. Rheb activates AMPK and reduces p27Kip1 levels in Tsc2-null cells via mTORC1-independent mechanisms: implications for cell proliferation and tumorigenesis. https://goo.gl/FdtacH
Dong Z, Liu Z, Cui P, Pincheira R, Yang Y, Liu J, Zhang JT. Role of eIF3a in regulating cell cycle progression. https://goo.gl/FTok1d
Pincheira R, Baerwald M, Dunbar JD, Donner DB. Sall2 is a novel p75NTR-interacting protein that links NGF signalling to cell cycle progression and neurite outgrowth. https://goo.gl/ejeyaJ
Pincheira R, Donner DB. The Sall2 transcription factor is a novel p75NTR binding protein that promotes the development and function of neurons. https://goo.gl/ka6OVr
Pincheira R, Castro AF, Ozes ON, Idumalla PS, Donner DB. Type 1 TNF receptor forms a complex with and uses Jak2 and c-Src to selectively engage signaling pathways that regulate transcription factor activity. https://goo.gl/5UKmK0
Satpathy M, Cao L, Pincheira R, Emerson R, Bigsby R, Nakshatri H, Matei D. Enhanced peritoneal ovarian tumor dissemination by tissue transglutaminase. https://goo.gl/lXwDBf
Gustin JA, Korgaonkar CK, Pincheira R, Li Q, Donner DB. Akt regulates basal and induced processing of NF-kappaB2 (p100) to p52. https://goo.gl/3k3Wud
Castro AF, Rebhun JF, Quilliam LA. Measuring Ras-family GTP levels in vivo--running hot and cold. https://goo.gl/iQefOu
Felekkis KN, Narsimhan RP, Near R, Castro AF, Zheng Y, Quilliam LA, Lerner A. AND-34 activates phosphatidylinositol 3-kinase and induces anti-estrogen resistance in a SH2 and GDP exchange factor-like domain-dependent manner. https://goo.gl/g0lxOV
Li Y, Asuri S, Rebhun JF, Castro AF, Paranavitana NC, Quilliam LA. The RAP1 guanine nucleotide exchange factor Epac2 couples cyclic AMP and Ras signals at the plasma membrane. https://goo.gl/J4r5OQ
Dong Z, Liu LH, Han B, Pincheira R, Zhang JT. Role of eIF3 p170 in controlling synthesis of ribonucleotide reductase M2 and cell growth. https://goo.gl/5VILoL
Gustin JA, Pincheira R, Mayo LD, Ozes ON, Kessler KM, Baerwald MR, Korgaonkar CK, Donner DB. Tumor necrosis factor activates CRE-binding protein through a p38 MAPK/MSK1 signaling pathway in endothelial cells. https://goo.gl/Jl0yVl
Gustin JA, Ozes ON, Akca H, Pincheira R, Mayo LD, Li Q, Guzman JR, Korgaonkar CK, Donner DB. Cell type-specific expression of the IkappaB kinases determines the significance of phosphatidylinositol 3-kinase/Akt signaling to NF-kappa B activation. https://goo.gl/VDLdHw
Castro AF, Rebhun JF, Clark GJ, Quilliam LA. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. https://goo.gl/1GNBpF
Wilkes DM, Wang C, Aristimuño PC, Castro AF, Altenberg GA. Nucleotide triphosphatase activity of the N-terminal nucleotide-binding domains of the multidrug resistance proteins P-glycoprotein and MRP1. https://goo.gl/lnuoZ3
Quilliam LA, Rebhun JF, Castro AF. A growing family of guanine nucleotide exchange factors is responsible for activation of Ras-family GTPases. https://goo.gl/1fYLmt
Pincheira R, Chen Q, Huang Z, Zhang JT. Two subcellular localizations of eIF3 p170 and its interaction with membrane-bound microfilaments: implications for alternative functions of p170. https://goo.gl/OGBPRm
Quilliam LA, Rebhun JF, Zong H, Castro AF. Analyses of M-Ras/R-Ras3 signaling and biology. https://goo.gl/vsZxBD
Pincheira R, Chen Q, Zhang JT. Identification of a 170-kDa protein over-expressed in lung cancers. https://goo.gl/9U0I9W
Mayo LD, Kessler KM, Pincheira R, Warren RS, Donner DB. Vascular endothelial cell growth factor activates CRE-binding protein by signaling through the KDR receptor tyrosine kinase. https://goo.gl/wPFW9L
Rebhun JF, Castro AF, Quilliam LA. Identification of guanine nucleotide exchange factors (GEFs) for the Rap1 GTPase. Regulation of MR-GEF by M-Ras-GTP interaction. https://goo.gl/wHWC4S
Burgos P, Metz C, Bull P, Pincheira R, Massardo L, Errázuriz C, Bono MR, Jacobelli S, González A. Increased expression of c-rel, from the NF-kappaB/Rel family, in T cells from patients with systemic lupus erythematosus. https://goo.gl/KxaOdo
Castro AF, Horton JK, Vanoye CG, Altenberg GA. Mechanism of inhibition of P-glycoprotein-mediated drug transport by protein kinase C blockers. https://goo.gl/8Tm0eG
Quilliam LA, Castro AF, Rogers-Graham KS, Martin CB, Der CJ, Bi C. M-Ras/R-Ras3, a transforming ras protein regulated by Sos1, GRF1, and p120 Ras GTPase-activating protein, interacts with the putative Ras effector AF6. https://goo.gl/3cN8tO
Vanoye CG, Castro AF, Pourcher T, Reuss L, Altenberg GA. Phosphorylation of P-glycoprotein by PKA and PKC modulates swelling-activated Cl- currents. https://goo.gl/kVkOuJ
Wang C, Castro AF, Wilkes DM, Altenberg GA. Expression and purification of the first nucleotide-binding domain and linker region of human multidrug resistance gene product: comparison of fusions to glutathione S-transferase, thioredoxin and maltose-binding protein. https://goo.gl/LIujII
Castro AF, Amorena C, Müller A, Ottaviano G, Tellez-Iñon MT, Taquini AC. Extracellular ATP and bradykinin increase cGMP in vascular endothelial cells via activation of PKC. https://goo.gl/T6ykP5
Wang G, Pincheira R, Zhang JT. Dissection of drug-binding-induced conformational changes in P-glycoprotein. https://goo.gl/7xftRG
Castro AF, Amorena C, Müller A, Ottaviano G, Tellez-Iñon MT, Taquini AC. Extracellular ATP and bradykinin increase cGMP in vascular endothelial cells via activation of PKC. https://goo.gl/RnYMkX
Castro AF, Altenberg GA. Inhibition of drug transport by genistein in multidrug-resistant cells expressing P-glycoprotein. https://goo.gl/T6tBH5
Horton JK, Thimmaiah KN, Altenberg GA, Castro AF, Germain GS, Gowda GK, Houghton PJ. Characterization of a novel bisacridone and comparison with PSC 833 as a potent and poorly reversible modulator of P-glycoprotein. https://goo.gl/l9G4xt
Wang G, Pincheira R, Zhang M, Zhang JT. Conformational changes of P-glycoprotein by nucleotide binding. https://goo.gl/u7pl7e
Amorena C, Castro AF. Control of proximal tubule acidification by the endothelium of the peritubular capillaries. https://goo.gl/SvPrPG
Amorena C, Castro A, Müller A, Villamil MF. Direct vascular effects in the rat of the vehicles used for the intravenous and oral administration of cyclosporin A. https://goo.gl/cybPS0